一、法规框架
进入欧盟的医疗器械必须满足以下三大指令:
• MDR 2017/745(原 MDD 93/42/EEC 升级版)
• IVDR 2017/746(原 IVDD 98/79/EC 升级版)
• AIMDD 90/385/EEC(有源植入器械)
产品符合任一指令后,方可加贴 CE 标志并在 27 个成员国自由流通。
三、八步流程
范围确认
核对产品预期用途,判断是否落入 MDR、IVDR 或 AIMDD。
基本要求对标
列出 GSPR(通用安全与性能要求)条款,形成差距清单。
协调标准选用
优先采用 EN ISO 13485、EN ISO 14971、EN 60601-1 等已公布的协调标准,以推定符合法规。
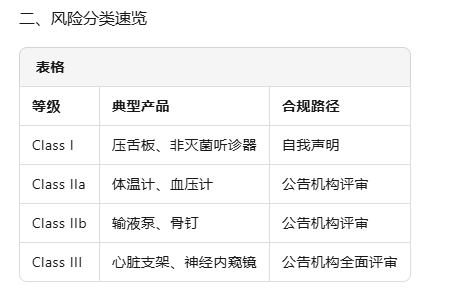
风险等级判定
依据 MDR 附录 VIII 规则,明确 I、IIa、IIb 或 III 类;一次性明确后续路径。
合规程序选择
• Annex II + III:全面质量管理体系
• Annex IX:EC 型式检验 + 生产质量保证
• Annex XI:产品验证
由企业规模、技术成熟度及公告机构能力综合决定。
技术文件与证据
建立技术文档(TD)与临床评价报告(CER);高风险器械需临床试验或等效性证明。
公告机构介入
仅 I 类(非无菌、非测量)可自我声明;其余须向欧盟 NANDO 名单内公告机构提交评审。
签发 CE 证书与 DOC
公告机构颁发 CE 证书后,企业拟定 EU 符合性声明(DoC),即可在产品、包装及说明书上加贴 CE 标志。
四、注意事项
• 证书有效期:通常为 5 年,年度监督审核不可缺席;
• 变更管理:设计、材料、预期用途变更须重新评估;
• UDI 与 EUDAMED:MDR 实施后,所有器械需注册唯一器械标识(UDI)并上传 EUDAMED 数据库。
总结
先分类、选标准、建体系、找公告机构,走完八步即可让医疗器械合法贴上 CE 标志,畅通欧盟市场。






 当前位置 -
当前位置 - 

